


Oxidant Defense in Normal and
Abnormal Red Blood Cells
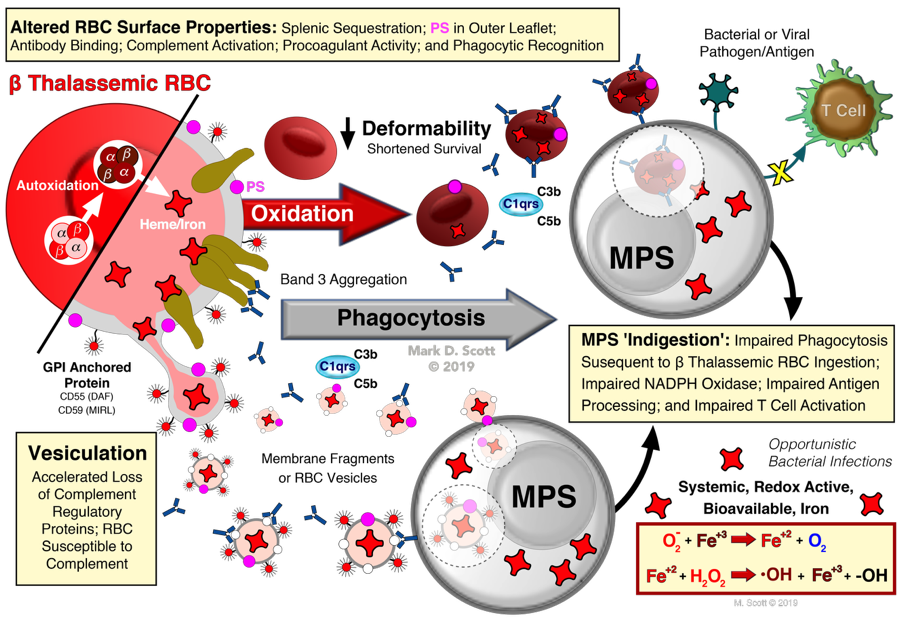
Schematically the pathophysiology of the β thalassemic RBC, and it's downstream consequences, as elucidated by the model human β thalassemic RBC, are summarized above. The initial autoxidation and subsequent GSH-dependent oxidation of the RBC also gives rise to membrane vesiculation of the thalassemic RBC. One consequence of membrane vesiculation is the preferential loss of phosphatidylinositol (PI) anchored proteins from the RBC. Among these PI-anchored proteins are decay accelerating factor (DAF; CD55) and the membrane inhibitor of reactive lysis (MIRL; CD59) both of which play important roles in preventing complement-mediated binding and lysis. The effects of the vesiculation-mediated loss of CD55 and CD59 can range from sublytic levels of bound complement enhancing phagocytosis to overt hemolysis. Indeed, a common endpoint for all the α-chain mediated injury is enhanced erythophagocytosis. As shown, oxidized RBC or the heme from these cells (Figure 8) significantly inhibits antigen processing, presentation and T cell proliferation. The systemic importance of this on cell-mediated immunity has not be fully appreciated and may potentially explain the predisposition of thalassemic patients to recurrent bacterial infections.
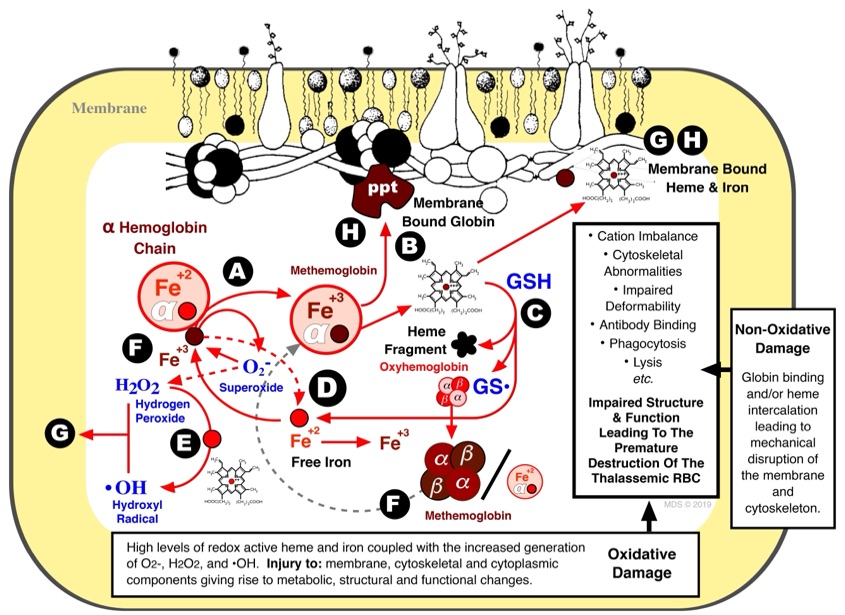
Containing approximately 20 mM iron, the RBC is the most ferruginous somatic cell in mammals. Under normal conditions, most of this iron is complexed within hemoglobin (as heme) with virtually none present as free metal (i.e., non-heme). This near perfect compartmentalization of iron may, however, break down in certain pathologic states such as β thalassemia and sickle cell disease resulting in the autoxidation of hemoglobin (i.e., formation of methemoglobin and hemichromes).
Our lab has extensively investigated the RBC pathophysiology in β thalassemia which is characterized by decreased/absent production of β hemoglobin chains and the presences of unpaired, highly unstable, α hemoglobin chains. Of physiologic importance, the monomeric α-chains spontaneously autoxidize to methemoglobin, simultaneously generating superoxide (O2-), at a rate 8-times that of normal hemoglobin (above). Moreover, consequent to the formation of methemoglobin, the non-covalently bound heme is more likely to escape the heme pocket of the globin chain giving rise to elevated levels of free, redox-active, intraerythrocytic iron. The O2- produced via the autoxidation of hemoglobin can reduce ferric (Fe+3) to ferrous (Fe+2) iron or form hydrogen peroxide (H2O2; either spontaneously or enzymatically via superoxide dismutase) . Importantly, the iron, O2-, and H2O2 can, via the Haber-Weiss Reaction, give rise to the formation of the 'dreaded' hydroxyl radical (•OH) which rapidly reacts with virtually all biological constituents converting pristine materials into biological garbage. Importantly, our studies with the human model β thalassemic RBC have demonstrated that the unpaired α-chains initiate an iron, GSH-dependent, self-amplifying and self-propagating reaction with the subsequent release of even more heme and, eventually, free iron. Membrane proteins and reactive thiol groups (not shown) were rapidly decreased in a pattern similar to that observed in vivo in β thalassemia.
Figures From:
Scott, M.D. Model Human ß Thalassemic Erythrocytes: Effect of unpaired purified α-hemoglobin chains on normal erythrocytes. In: Beta Thalassemia (Editor: Zakaria, M.), INTECH. Croatia. ISBN: 978-1-83880-587-6 (2019). In press. DOI: 10.5772/intechopen.90288